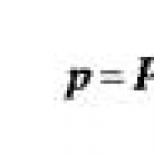Stargardt'ın göz hastalığı tedavisi. Stargardt hastalığının ayırıcı tanısının modern olanakları. Stargardt hastalığının tedavisi ve prognozu
Santral sinir sisteminin klasik bir örneği olan Stargardt hastalığı pigment dejenerasyonu, 20. yüzyılın başında K. Stargardt (1909, 1913) tarafından tanımlanmıştır. çocukluk ve genç yaşta (7-20 yaş) kendini gösteren maküler bölgenin kalıtsal bir hastalığı olarak. Fundustaki değişiklikler, polimorfik olmasına rağmen, her iki gözde de pigmente yuvarlak noktaların görünümü, depigmentasyon alanları ve retina atrofisi ile karakterizedir. pigment epiteli(RPE), bazı durumlarda "boğa gözü" tipinde, genellikle paramaküler bölgede beyazımsı-sarımsı noktalarla birleşir. Çocuklarda progresif maküler dejenerasyonun benzer bir klinik tablosu 19. yüzyılın başlarında tanımlanmıştır.
Sarımsı-beyazımsı noktalar ve şeritler şeklindeki değişiklikler, makula bölgesinde değişiklik olsun veya olmasın A. Franceschetti, "fundus flavimaculatus" terimini belirledi. Literatürde, "Stargardt hastalığı" ve "fundus flavimaculatus" terimleri sıklıkla birleştirilir (Stargardt hastalığı / fundus flavimaculatus), böylece varsayılan köken birliği ve / veya hastalığın bir biçiminden (Stargardt hastalığı) diğerine geçişi vurgular. (fundus flavimaculatus) geliştikçe.
Tipik nedeniyle görme bozukluğu varsa distrofik değişiklikler makula, yaşamın ilk yirmi yılında başlar, "Stargardt hastalığı" terimini kullanmak tercih edilir. İleri yaşlarda retinanın orta ve periferik kısımlarında değişiklikler meydana geliyorsa ve hastalık daha akut ilerliyorsa "fundus flavimaculatus" teriminin kullanılması önerilir.
 Bunun kalıtsal geçişli heterojen bir hastalık grubu olduğu tespit edilmiştir.
Bunun kalıtsal geçişli heterojen bir hastalık grubu olduğu tespit edilmiştir.
Belirtiler (görünüş sırasına göre):
- Foveada - değişiklik olmadan veya pigmentin yeniden dağıtılmasıyla
- Beyaz-sarı lekelerle çevrili olabilen "salyangoz izi" tipi veya bronz refleksin oval lezyonları.
- "Coğrafi" atrofi, "boğa gözü" gibi görünebilir.
sınıflandırma
Fundus flavimaculatus olan ve olmayan maküler distrofi dahil olmak üzere iki tip Stadgardt hastalığı arasındaki klasik ayrımın yanı sıra, varyasyonlara dayalı birkaç başka sınıflandırma önerilmiştir. klinik tablo göz fundusu.
Evet, K.G. Soylu ve R.E. Carr (1971) dört tür hastalık tanımlamıştır:
- Tip I - beneksiz makula dejenerasyonu (benekli). Görme keskinliği erken azalır.
- II - parafoveal benekli,
- III - yaygın benekli maküler dejenerasyon,
- Tip IV - makula dejenerasyonu olmadan yaygın beneklenme. Retina lezyonu foveal bölgeyi etkilemediği için görme keskinliği oldukça yüksek kalır.
genetik araştırma
Stargardt distrofisi en yaygın olarak otozomal resesif bir şekilde kalıtılır, ancak hastalığın otozomal baskın bir şekilde bulaştığı birçok aile tanımlanmıştır. Baskın kalıtım tipinin, esas olarak Stargardt hastalığının III ve IV tiplerinin özelliği olduğuna dair bir görüş vardır.
 Konumsal klonlama ile belirlenen yer hastalığa neden olan ABCR olarak adlandırılan, fotoreseptörlerde eksprese edilen Stargardt hastalığı geni. ABCR'nin diziliminde insan RmP geni ile aynı olduğu gösterilmiştir.
Konumsal klonlama ile belirlenen yer hastalığa neden olan ABCR olarak adlandırılan, fotoreseptörlerde eksprese edilen Stargardt hastalığı geni. ABCR'nin diziliminde insan RmP geni ile aynı olduğu gösterilmiştir.
RmP proteini, görsel hücrelerin dış bölümlerinin disklerinin kenarında lokalize olan, moleküler ağırlığı 210 kDa olan bir integral membran glikoproteinidir. RmP'nin, ATP hidrolizini uyaran ve hücre zarları boyunca spesifik substratların ATP'ye bağlı hareketini etkileyen ATP bağlayıcı kasetlerin ABC taşıyıcılarının üst ailesine ait olduğu gösterilmiştir.
ABC taşıyıcılarının üst ailesinin birkaç üyesi için genlerin, insan retinasının bir dizi kalıtsal hastalığının gelişiminde rol oynadığı bulundu. Böylece, Stargardt hastalığının otozomal dominant kalıtım tipinde, mutasyona uğramış genlerin 13q ve 6ql4 kromozomları üzerindeki lokalizasyonu gösterildi ve Stargardt benzeri retina hastalığının (muhtemelen tip IV ile ilişkili) yeni bir baskın formu için bir gen haritalandı. D4S1582 ve D4S2397 belirteçleri arasında kromozom 4p.
İnsan RmP geni, kromozom lp (Ip21-pl3) üzerindeki D1S424 ve D1S236 belirteçleri arasında eşlenir. Stargardt distrofisi ve fundus flavimaculatus'un en yaygın otozomal resesif formunun genleri de burada lokalizedir ve otozomal resesif retinitis pigmentosa formu RP19 geninin yeri, lp kromozomu üzerindeki D1S435-D1S236 belirteçleri arasında belirlenir. S.M. tarafından yapılan bir çalışmada. Azaryan et al. (1998), ABCR geninin tam ince intron-ekson yapısını oluşturdu.
İmmünofloresan mikroskopisi ve Western blot analizi kullanılarak, ABCR'nin foveal ve perifoveal konilerde mevcut olduğu gösterildi ve bu nedenle Stargardt distrofisindeki merkezi görme kaybının, ABCR'deki mutasyonların neden olduğu foveal koni dejenerasyonunun doğrudan bir sonucu olabileceğine inanılıyor. gen.
Ayrıca, ABCR mutasyonlarının, eksüdatif olmayan yaşa bağlı makula dejenerasyonu (AMD) ve koni-çubuk dejenerasyonu olan hastaların bir alt popülasyonunda mevcut olduğu ortaya çıktı; bu, Stargardt'lı hastaların akrabalarında genetik olarak belirlenmiş bir AMD geliştirme riskinin varlığını düşündürür. hastalık. Bununla birlikte, Stargardt hastalığı ve AMD'nin fenotipik ve genotipik belirtilerinin ABCR genindeki mutasyonlarla ilişkili olduğuna şüphe olmamasına rağmen, bu ifade tüm araştırmacılar tarafından desteklenmemektedir.
J.M. Roset et al. (1999), üyeleri arasında hem retinitis pigmentosa hem de Stargardt hastalığı olan hastaları olan bir aileyi inceleyerek, ABCR geninin heterozigotluğunun Stargardt distrofisi gelişimine ve homozigotluğun retinitis pigmentosa gelişimine yol açtığını göstermiştir.
Bu nedenle, son genetik çalışmaların sonuçları, retinitis pigmentosa, Stargardt hastalığı, fundus flavimaculatus ve AMD'nin klinik tablosundaki açık farklılıklara rağmen, bunların ABCR lokusunun alelik bozuklukları olduğunu göstermektedir.
Stargardt distrofisinin geniş fenotipik belirtileri ve tespit yaşı klinik işaretler(yaşamın ilk ila yedinci on yılına kadar), bir ailede bile gözlenmesi, görme keskinliğindeki değişiklikleri ayırt etmeyi ve tahmin etmeyi zorlaştırır. Anjiyografi verileri, tıbbi öykü, azalmış görsel fonksiyon, ERG'deki değiştirilmiş koni bileşenleri, lokal ve multifokal ERG'deki spesifik değişiklikler tanı koymaya yardımcı olur.
Böylece, son yıllar genetik araştırmaların sonuçlarına giderek daha fazla önem verilmektedir. Evet, G.A. Fishman et al. (1999), ABCR gen mutasyonlarına sahip Stargardt distrofisi ve fundus flavimaculatus'lu geniş bir hasta grubunu inceleyerek, fenotipik belirtilerin değişkenliğinin belirli bir şekilde spesifik amino asit dizisinin varyasyonuna bağlı olduğunu gösterdi. Floresein anjiyografi, oftalmoskopi, elektroretinografik ve perimetrik çalışmaların sonuçlarına göre tanımlamışlardır. üç hastalık fenotipi
- Bu fenotiplerden biri, makula atrofik lezyonları ile birlikte perifoveal sarımsı beyaz lekelerin ortaya çıkması, koyu bir koroidin olmaması ve ERG dalgalarının normal genliği ile karakterize edilir. Bu fenotipte, ABCR geninin ekson 42'sindeki dizide, glisinin glutamin (Gly]961Glu) ile yer değiştirmesinden oluşan bir değişiklik bulundu.
- Başka bir fenotip, koyu bir koroid ve fundus üzerinde daha dağınık bir şekilde dağılmış sarımsı beyaz noktalar ile karakterize edildi, ancak hiçbir Glyl961Glu ikamesi saptanmadı.
- RPE'de ciddi atrofik değişiklikler ve azalmış çubuk ve koni ERG'si olan fenotipte, ABCR mutasyonu 7 hastadan sadece birinde bulundu.
ABCR mutasyonlarına çeşitli fenotipik belirtilerin eşlik etmesi nedeniyle, spesifik gen mutasyonları ile klinik fenotipler arasındaki korelasyonların belirlenmesindeki ilerlemelerin, hastalara ve görme keskinliğinin prognozu hakkında bilgi verilmesini kolaylaştıracağına inanılmaktadır.
Tüm bu çalışmalar, yalnızca retinanın genetik hastalıklarının ince mekanizmalarını çözmeyi değil, aynı zamanda onlar için olası bir tedavi bulmayı da amaçlıyor.
Klinik tablo
 Görüş Hattı
Görüş Hattı
Fundus flavimaculatus ile, özellikle yaşamın ilk yirmi yılında görüş alanı değişmeyebilir; Stargardt hastalığı olan tüm hastalarda, sürecin makuladaki yayılmasına bağlı olarak çeşitli büyüklüklerde rölatif veya mutlak santral skotomlar saptanır. bölge.
renkli görüş
Stargardt hastalığı tip I olan hastaların çoğunda döteranopi vardır; tip II Stargardt hastalığında renk görme bozuklukları daha belirgindir ve sınıflandırılamaz. Renk anomalisinin tipi, patolojik süreçte ağırlıklı olarak hangi tip konilerin yer aldığına bağlı gibi görünmektedir, bu nedenle, fundus flavimaculatus ile renk görüşü değişmeyebilir veya kırmızı-yeşil dikromazi not edilebilir.
Karanlık adaptasyon
O. Gelişken'e göre, J.J. De Jaey (1985), Stargardt hastalığı ve fundus flavimaculatus'u olan 43 hastadan 4'ünde yüksek ışık duyarlılığı eşiği vardı, 10'unda karanlık adaptasyon eğrisinin koni segmenti yoktu.
Mekansal kontrast duyarlılığı
Stargardt distrofisi ile, orta uzamsal frekanslar bölgesinde önemli bir düşüş ve yüksek uzamsal frekanslar bölgesinde tamamen yokluğu ile tüm frekans aralığında değiştirilir - bir koni distrofisi paterni.
Kontrast duyarlılığı Arka plandan daha koyu ve daha açık bir uyaranın sunulması üzerine sensorimotor reaksiyonun zamanı ile tahmin edilen koni sisteminin açık ve kapalı aktivitesi, retinanın orta bölgesinde yoktur ve bir miktar off-duyarlılık korunur. merkezden 10°'lik bölge.
Elektroretinografi ve elektrookülografi
 Elektrofizyolojik yöntemlerden, retinanın maküler bölgesinin hastalıklarının tanı ve ayırıcı tanısında elektroretinografi ve elektrookülografi en bilgilendiricidir.
Elektrofizyolojik yöntemlerden, retinanın maküler bölgesinin hastalıklarının tanı ve ayırıcı tanısında elektroretinografi ve elektrookülografi en bilgilendiricidir.
Literatüre göre, Stargardt distrofisi ve fundus flavimaculatus, general veya ganzfeld'in başlangıç evrelerinde ERG normaldir. Bununla birlikte, çeşitli metodolojik elektroretinografi yöntemlerinin kullanılması konuyu değerlendirmemize izin verir. fonksiyonel bozukluklar retinada çeşitli katmanları ve bölümleri düzeyinde.
Bu nedenle, bir vantuz lensine monte edilmiş bir LED kullanarak yerel bir ERG'yi (LERG) kaydederken, makula bölgesinin biyopotansiyelleri zaten normalin altındadır. İlk aşama ganzfeld-ERG'nin normal genliklerinin aksine Stargardt distrofisi. Süreç ilerledikçe LERG tamamen yok olana kadar azalır. Diğer yazarlar ayrıca en yüksek gecikmede bir artışa ve yerel foveal yanıtların genliklerinde bir azalmaya dikkat çeker; görme keskinliği 20/20 - 20/30 olan fundus flavimaculatus hastalarının %64'ünde.
Zonal elektroretinografi kullanımı, Stargardt hastalığının erken evrelerinde retinanın dış tabakasının (fotoreseptörler) reaksiyonunun sadece makula bölgesinde değil, aynı zamanda paramaküler ve periferik bölgelerde de inhibisyonunu ortaya çıkarırken, retinanın proksimal tabakaları korunmuşlardı.
Retinanın farklı bölgelerinde (merkez, paramerkez, çevre) a- ve 1a ERG dalgalarının genliklerinde bir azalma, zaten hastalığın ilk aşamasında her iki sistemin (koni ve çubuk) tüm fotoreseptör tabakasının genelleştirilmiş bir lezyonunu gösterir. . Sürecin gelişimine yayılma eşlik ediyor patolojik değişiklikler ERG'nin tüm bileşenlerinde algılama sıklığında ve değişikliklerin ciddiyetinde bir artış olarak ifade edilen retinanın derinliklerinde.
Bununla birlikte, zaten Stargardt hastalığının ilk (I-II) aşamalarında, ERG'nin koni bileşenlerinin çubuk olanlara kıyasla daha yüksek derecede inhibisyonu ortaya çıkar.
P. A. Blacharski'ye (1988) göre, uzun süreli karanlık adaptasyondan (45 dakika) sonra, fundus flavimaculatuslu hastalarda, ERG'nin fotopik bileşenlerinde sağlıklı bireylere göre daha büyük (% 29) bir azalma kaydedilmiştir. Skotopik ERG'nin yanıtları sadece %6-10 oranında hafifçe azalır. J.B.M. Moloney et al. (1983), incelenenlerin %100'ünde koni ERG'sinde bir artış ve %50'sinde çubuk ERG'de bir azalma tespit etmiştir.
R. Itabashi ve ark. (1993), çeşitli ERG bileşenlerinin inhibisyon derecesini karşılaştıran, Stargardt hastalığı olan geniş bir hasta grubu üzerinde yapılan bir araştırmanın sonuçlarını sunmuştur.
K.G. tarafından önerilen sınıflandırmaya göre. Soylu ve R.E. Sugg (1971), hastalığın evrelerine göre birkaç hasta grubu ayırdı: 1-4. Tüm ERG bileşenlerinin ortalama genlikleri, retina koni sisteminde daha belirgin değişikliklerle normal değerlerin altında çıktı. Fotopik b-dalgası normun %57.4'ü, skotopik b-dalgası - %77.9'u, "beyaz" titreşen uyarana verilen tepkiler 32 Hz - %78.9, a-dalgası - %87.7'si, b-dalgası - normun %95,8'i idi. Tüm ERG bileşenlerinde en büyük düşüş grup 3'teki hastalarda gözlendi.
Zamanlama parametreleri de değiştirildi; zirve süresinin uzaması, özellikle grup 3 hastalarında, a-dalgası için en önemlidir. Bu aşama aynı zamanda, normalin altında bir aydınlık-karanlık EOG katsayısının (%73.5) en sık saptanmasıyla da karakterize edilir. Yazarlara göre, grup 3'teki hastalar için prognoz en olumsuz olanıdır.
7-14 yıl boyunca hastaların gözlemlenmesi, klinik süreçle karşılaştırıldığında elektrofizyolojik parametrelerin dinamiklerini izlemeyi mümkün kılmıştır. Daha belirgin oftalmoskopik değişikliklere hem elektroretinografik hem de elektrookülografik parametrelerde bir azalma eşlik etti. Bu sonuçlar, elektroretinografik ve histolojik verilere dayanarak, fundus flavimaculatus'ta RPE'de bir başlangıç lezyonu ve Stargardt distrofisinde retinal fotoreseptörlerin daha ileri bir lezyonunu öneren diğer araştırmacıların görüşleri ile tutarlıdır.
Literatürde elektrookülografi sonuçlarında bazı farklılıklar vardır. Çoğu zaman, fundus flavimaculatus ve Stargardt distrofisi olan hastaların çoğunda normal veya hafifçe azalmış bir EOG not edilir. Bununla birlikte, bir dizi araştırmacı, Arden katsayısı açısından yüksek bir normal altı EOG yüzdesine dikkat çekiyor: FF'li hastaların %75-80'inde. Çoğu yayının, birkaç hasta grubuna yönelik bir anketin sonuçlarını sunduğu dikkate alınmalıdır: 3 ila 29.
G.A. Fishman (1976, 1979) fundus flavimaculatus evrelerini EOG sonuçlarıyla ilişkilendirdi. Evre I-II hastalığında, muayene edilen tüm hastalarda EOG'nin değişmediğini (28/28), evre III-IV'te ise hastaların %90'ında normalin altında olduğunu gösterdi. G.A.'ya göre Fishman ve arkadaşları (1976 1977 1979), ancak patolojik süreç retinanın önemli bir alanını etkiliyorsa, EOG anormal olacaktır. Diğer araştırmacılar da fundus flavimaculatus'lu hastaların büyük çoğunluğunda EOG değişikliklerinin olmadığına dikkat çekiyor. Çalışmaların sonuçlarının farklılıklarından etkilenmesi olasıdır. metodolojik teknikler standartlaştırma girişimlerine rağmen.
Bu nedenle, elektrofizyolojik çalışmaların, Stargardt hastalığı ve fundus flavimaculatus'un ayırıcı tanısında yardımcı olmaktan ziyade, retinanın koni ve çubuk sistemlerindeki değişikliklerin varlığını ve şiddetini ortaya koyması ve RPE'nin durumunu değerlendirmesi daha olasıdır.
Ayırıcı tanı
Bazı kalıtsal hastalıklarda klinik tablo Stargardt hastalığındakine benzer olabilir. Bu tür hastalıklar arasında baskın ilerleyici foveal distrofi, koni-çubuk ve çubuk-koni (retinitis pigmentosa) distrofisi, jüvenil retinoskizi bulunur. Atrofik maküler dejenerasyon, oligopontoserebral atrofi dahil olmak üzere çeşitli spinoserebral ve serebral spastik bozukluklarda tanımlanmıştır. Benzer morfolojik bulgular, klorokin retinopatisi veya hamile kadınların şiddetli toksikozunun oküler belirtileri gibi kalıtsal olmayan hastalıklarda da tanımlanmıştır.

Fundus resmindeki farklılıklar, yaş, hastalığın başlangıcı, fonksiyonel araştırma yöntemlerinden elde edilen veriler temelinde, S. Merin (1993) iki ana Stargardt hastalığı türü tanımladı.
Stargardt hastalığı tip I
Bu tip en çok orijinal olarak tanımlanan Stargardt hastalığı ile uyumludur. Bu, klinik belirtileri çocuklarda 6-12 yaşlarında gözlenen genç kalıtsal makula dejenerasyonudur. Erkekler ve kızlar aynı sıklıkta hastalanırlar, kalıtsal geçiş otozomal çekinik tipe göre gerçekleştirilir.
Hastalık iki taraflı ve simetrik olarak kendini gösterir. İleri aşamalarda, foveal refleks yoktur. Retina pigment epiteli (RPE) seviyesindeki değişiklikler, hiper ve depigmentasyon alanlarıyla çevrili kahverengimsi bir pigmentin merkezinde bir birikim olarak görünür. Klinik tablo bir "boğa gözüne" benziyor.
Floresein anjiyografi, tipik "boğa gözü" fenomenini doğrular. Karanlık, floresan geçirmeyen merkez, geniş bir hipofloresan nokta halkasıyla çevrilidir ve bunu genellikle başka bir hiperpigmentasyon halkası izler. Bu patern, fundus merkezi bölgesindeki pigment miktarındaki artış, komşu RPE hücrelerinin atrofisi ve pigment epitelinin atrofi ve hipertrofisinin bir kombinasyonu ile açıklanmaktadır. Makula bölgesinde floreseinin olmaması "sessiz koroid" veya koyu koroid olarak adlandırılır ve RPE'de asit mukopolisakkaritlerin birikmesi ile açıklanır. D.A.'ya göre Klein ve A.E. Krill (1967), "boğa gözü" fenomeni, tip I Stargardt hastalığı olan hemen hemen tüm hastalarda tespit edilir.
Hastalık ilerledikçe görme keskinliği azalır ve görme azlığına neden olur. Hastalığın erken evrelerinde ERG ve EOG ileri evrelerde normal kalırsa, koni sisteminin ERG verilerine göre tepkileri azalır ve EOG göstergeleri orta derecede normalin altına düşer. Hastalarda ağırlıklı olarak koni sisteminin yenilgisi ile bağlantılı olarak, daha sık olarak deuteranopia tipine göre renk görme de bozulur.
Bir araba kazası sonucu ölen tipik Stargardt hastalığı tip I olan bir hastanın iki gözünün histolojik incelemesinde, R.C. Kartal ve ark. (1980), RPE hücrelerinin boyutunda önemli bir değişkenlik buldu - 14 ila 83 mikron. Büyük RPE hücreleri, ultrastrüktür, otofloresan ve histokimyasal özellikler açısından patolojik (anormal) lipofusine karşılık gelen granüler bir madde oluşturdu. Melanin miktarı azaltıldı ve melanin granülleri hücrenin içine doğru kaydırıldı.
Daha fazlası geç aşamalar Stargardt hastalığı, retinanın maküler bölgesinden fotoreseptörlerin ve RPE hücrelerinin çoğunun kaybolduğunu ortaya çıkardı. Aynı zamanda bazı RPE hücreleri lipofuscin birikimi ile dejenerasyon aşamasındaydı, atrofi alanlarının kenarlarında RPE hücrelerinin hiperplazisi gözlendi.
F. Schutt ve ark. (2000), Stargardt hastalığı, AMD ve retina yaşlanması dahil olmak üzere yoğun lipofusin birikimi ile ilişkili retina hastalıklarında, lipofuscin A2-E'nin (N-retiniliden-N-retinil-etanol-amin) retinoid floresan bileşeninin olduğunu göstermiştir. Lizozomların parçalayıcı işlevini zayıflatır ve RPE hücrelerinin intralizozomal pH'ını yükselterek membran bütünlüğünün kaybolmasına neden olur. Lizozomotropik özelliklere ek olarak, A2-E'nin fotoreaktif özellikleri ve fototoksisitesi gösterilmektedir.
Stargardt hastalığı tip II
Tip I'den farklı olarak retinanın makula bölgesindeki tipik değişikliklerin yanı sıra fundus üzerinde ekvatora kadar ulaşabilen çoklu ve yaygın FF noktaları vardır. Tip II Stargardt hastalığında görme keskinliğindeki azalmanın daha yavaş gerçekleşmesi ve sonuç olarak hastaların daha sonra göz doktoruna başvurması nedeniyle olsa da hastalık biraz daha geç başlar. Stargardt hastalığı tip II'de maküler bölgenin sınırlarını aşan daha fazla değişiklik olması nedeniyle, elektrofizyolojik veriler tip I'dekilerden farklıdır.
Böylece, ERG'de çubuk sisteminin tepkileri önemli ölçüde azalır. EOG parametreleri de değişti daha fazla. Sarımsı lekelerin makula bölgesi (makula lutea) dışında yüksek oranda vakada bulunması, Stargardt hastalığı ile FF arasında net bir ayrım yapmayı zorlaştırır.
Fundus flavimaculatus
Kural olarak, fundus flavimaculatus veya sarı benekli fundus, Stargardt hastalığı ile birleştirilir ve izole bir retina hastalığı şekli olarak yaygın değildir. Tipik ("temiz") vakalarda, hastalarda neredeyse hiçbir hastalık belirtisi görülmez. Görme keskinliği, renkli görme, görüş alanı normal sınırlar içindedir. Karanlık adaptasyon normal veya biraz azaltılmış olabilir. Fundusta, makula ve retinanın çevresi değişmez, sadece fovea ve ekvator arasında çok sayıda grimsi veya sarımsı noktalar görülebilir çeşitli şekiller: yuvarlak, oval, uzun, virgül veya balık kuyruğu şeklinde, birleşebilen veya birbirinden ayrı yerleştirilebilen, küçük - 200-300 mikron veya 3-5 kat daha büyük. Dinamik gözlem altında bu lekelerin rengi, şekli, boyutu değişebilir. Başlangıçta sarımsı ve iyi tanımlanmış noktalar, belirsiz sınırlarla gri hale gelebilir veya birkaç yıl sonra kaybolabilir.
Paralel olarak, floresan anjiyografi ile ortaya çıkan resim farklılaşıyor: hiperfloresanlı alanlar hipofloresan hale geliyor. Hastalığın gelişiminin sonraki aşamalarında, RPE atrofisi, bireysel noktaların kaybolması ve bunların düzensiz hipofloresan alanları ile değiştirilmesi olarak kendini gösterir.
Fundus flavimaculatus'taki (FF) lekelerdeki benzer değişiklikler, her iki Stargardt hastalığı tipinin özelliğidir, ancak FF'nin "saf formunda" daha az belirgindirler.
Hastalığın başlangıcı ve büyük olasılıkla tespit zamanı yaşa bağlı değildir. FF'nin otozomal resesif bir kalıtım türü olduğu varsayılır, ancak bazı durumlarda bu patolojinin kalıtsal yapısını belirlemek mümkün değildir.
Stargardt'ın makula dejenerasyonu(Stargardt maküler distrofisi, STGD) - en yaygın kalıtsal maküler dejenerasyon, oluşumu 10.000'de 1'dir; hastalık otozomal resesif bir şekilde kalıtılır. Çoğu vaka, yaşamın ikinci on yılının başında merkezi görüşte bir azalma ile kendini gösterir. Maküler atrofi genellikle gözün arka kutbunda retina pigment epiteli (RPE) seviyesinde sarı-beyaz pul pul tortularla gelişir.
Tortular yuvarlak, oval, lunat veya balık benzeri (pisciform) olabilir. Erken evrelerde maküler atrofinin oval bölgesi "dövme bronz" gibi görünebilir. Bununla birlikte, hastalığın erken döneminde pul pul tortular olmayabilir ve tek anormallik maküler atrofi olabilir; ancak, kural olarak, mevduat daha sonra görünür. Sarı noktalı bir fundus (fundus flavimaculatus, FFM) paterni, maküler atrofi yokluğunda pul pul tortuların görünümü ile gelişir.
VE sarı benekli fundus aynı genin mutasyonlarından kaynaklanan; aynı ailede her iki tür değişiklik de gözlenebilir. Sarı benekli fundusu olan hastaların çoğunda daha sonra maküler atrofi gelişir.
ve Stargardt hastalığı, ve sarı benekli fundus erken fazda floresan anjiyografide, klasik olarak karanlık veya gizli koroid. Bunun nedeni, koroid kılcal damarlarının floresansının taranmasının bir sonucu olarak retina pigment epiteli tarafından aşırı lipofusin birikmesidir. FA üzerindeki gelişimlerinin erken evrelerinde flokülent retinal birikintiler hipofloresan görünür, ancak daha sonra retina pigment epitelinin atrofisi nedeniyle hiperfloresan hale gelirler.
Teşhisi doğrulamak için, FAG'a alternatif olarak, retina pigment epitelinin lipofussinin floresansının sabitlenmesine dayanan bir otofloresan çalışması yapılır. Anormal lipofuscin birikimi, aktif ve emilebilir flokülent tortuların varlığı ve RPE atrofisi - özellikler otofloresan çalışmasında tespit edildi. Makula disfonksiyonu ve fundusta değişiklik olmaması nedeniyle görme bozukluğu olan çocuklarda FAG hala bilgilendiricidir; makula bölgesinin merkezinde göze çarpmayan pencereli bir kusur veya koyu renkli bir koroid tanıyı doğrulamaya yardımcı olur.
saat optik koherens tomografi(OCT), makula bölgesinin periferik bölgesinin yapısının göreceli olarak korunmasıyla, maküler bölgenin merkezi bölgesinin retinasının dış katmanlarının arkitektoniğinin genellikle bir kaybını veya belirgin bir ihlalini ortaya çıkarır.
Arka kutbun retina pigment epiteli seviyesinde sarı-beyaz pul pul birikintiler.
Erken başlangıçlı maküler atrofi.
B) elektrofizyoloji. Stargardt hastalığındaki elektrofizyolojik değişiklikler değişkendir. Retina pigment epitelinin genel bir işlev bozukluğunu gösteren anormal bir elektrookülogram (EOG) sıklıkla kaydedilir. Model elektroretinogram (PERG) ve fokal elektroretinogram genellikle soluktur veya amplitüdü belirgin şekilde azalır, bu da maküler disfonksiyonu düşündürür. Tanı anında Ganzfeld-ERG değişmeyebilir (grup 1) veya geniş retina hasarına işaret edebilir (grup 2 ve 3):
Grup 1: normal ganzfeld ERG ile şiddetli patern ERG anormallikleri.
Grup 2; ek olarak genelleştirilmiş koni disfonksiyonu.
Grup 3: koni ve çubukların genel disfonksiyonu.
Bu gruplar, hastalığın başlangıç yaşına veya süresine bağlı değildir; elektrofizyolojik gruplar farklı fenotipik alt tipleri temsil edebilir ve bu nedenle bir prognoz yapmada bilgilendirici olabilir. Birinci grubun hastaları daha yüksek görme keskinliğine, daha sınırlı flokülent birikinti dağılım alanlarına ve maküler atrofiye sahiptir; üçüncü grubun hastalarında görme keskinliğinde daha şiddetli bir azalma, daha geniş bir topaklanma dağılımı alanı ve toplam maküler atrofi vardır.
içinde) Moleküler genetik ve patogenez. Stargardt hastalığı / sarı benekli fundusun patogenezi, ABCA4 genindeki, ayrıca retinitis pigmentosa ve koni-rod distrofisinin gelişmesine neden olan mutasyonlara dayanmaktadır. ABCA4, retinoidlerin fotoreseptörden retina pigment epiteline taşınmasında rol oynayan, çubukların ve konilerin dış bölümlerinin disklerinin bir transmembran kenar proteinini kodlar. Bu taşımadaki bir kusur, retina pigment epitelinde florofor lipofuskin, A2E (N-retiniliden-N-retniletanolamin) birikmesine yol açar, bu da ölümüne neden olur ve ikincil fotoreseptör dejenerasyonuna yol açar.
ABCA4 sekansının 500'den fazla varyantı, yüksek allelik heterojenite gösteren tarif edilmiştir; sonuç olarak, bu kadar büyük (50 ekzon) bir polimorfik genin patojenik dizisinin tanımlanması önemli zorluklara neden olur. Kodlanmış protein üzerinde belirgin bir etkiye sahip olan anlamsız mutasyonların patojenik olacağı güvenle tahmin edilebilir. Yanlış anlamlı mutasyonları analiz ederken, bu tür dizi varyantları sıklıkla kontrol numunelerinde meydana geldiğinden büyük zorluklar ortaya çıkar; sonuç olarak, tanımlanan mutasyonun patojenitesini doğrulamak çok sorunlu olabilir.
Patojenitenin doğrudan doğrulanması, yalnızca mutant gen tarafından kodlanan proteinin fonksiyonel analizi ile elde edilebilir. Stargardt hastalığında, en sık olarak ABCA4 Gly-1961Glu genindeki bir mutasyon tespit edilir; Ala1038Val mutasyonu da yaygındır.
ABCA4 mutasyonlarının tipi ve kombinasyonu ile fenotipik belirtilerin şiddeti arasında bir korelasyon kurmak çoğu zaman mümkündür. Örneğin, bialelik boş mutasyonlar genellikle Stargardt hastalığından ziyade bir koni-çubuk distrofisi fenotipine neden olur. Retinadaki fenotipik değişikliklerin değişkenliği açıklanmıştır çeşitli kombinasyonlar Aynı aile içinde meydana gelen ABCA4 mutasyonları; ek değiştirici genlerin veya çevresel faktörlerin de aile içi değişkenliği etkilemesi muhtemeldir.
A2E dahil olmak üzere lipofuscin metabolik ürünlerinin birikimi Stargardt hastalığında ve ABCA4 nakavt farelerde (abca4-/-) gözlenir; bu, serbest radikallerin oluşumuna, proapoptotik mitokondriyal proteinlerin salınımına ve lizozom disfonksiyonuna yol açar. Sonuç olarak, retina pigment epitel hücrelerinin işlev bozukluğu ve ölümü gelişir, bu da fotoreseptörlerin ölümüne yol açar.
abca4-/-- fareler tamamen karanlıkta bırakıldığında A2E sentezi yavaşlar ve yiyeceklerine A vitamini eklendiğinde hızlanır.Stargardt hastalığı olan hastaların ek A vitamini alımından kaçınmalarını ve koyu renkli kullanmalarını önermek mantıklı görünmektedir. Güneş gözlüğü UV filtreleri ile. Ayrıca retina distrofilerinin hayvan modellerinde fotoreseptör ölümünü yavaşlatan antioksidanlar açısından zengin bir diyet öneriyoruz. Hasta çocukların az görme ve eğitim desteği konusunda yardıma ihtiyacı olabilir.
Bir hastada hasta çocuk sahibi olma riski %1'dir (hastanın eşi yakın akrabası ise bu olasılık artar). Stargardt hastalığının taşıyıcı sıklığı 50'de 1'dir; Bir partnerin, patojenik olarak değiştirilmiş bir ABCA4 gen dizisinin asemptomatik bir taşıyıcısı olma şansı 50'de 1'dir.
G) Umut verici terapi alanları. Stargardt hastalığının tedavisine yönelik yeni terapötik yaklaşımlar, ATP'ye bağlı olarak hareket eden ilaçları içerir. taşıma mekanizması ve böylece ABCA4'e bağlı retinoid taşınmasını hızlandırır veya A2E üretimini azaltarak görsel döngüyü yavaşlatır. A2E'nin toksik etkilerini doğrudan engellemek daha etkili olabilir. Bu üç alanın her birinde hareket eden ilaçlar geliştirilmiştir; yakın gelecekte insan klinik deneylerinin yapılması muhtemeldir. Benzer ilaçlar, Best hastalığı gibi lipofuskin birikiminin eşlik ettiği diğer makula dejenerasyonlarının tedavisinde etkili olabilir.
Diğer tedavi yolları, sırasıyla büyüme faktörlerini veya transplant retinal pigment epitelyum/fotoreseptör hücrelerini artırmak için gen takviyesi, hücre tedavisi veya kök hücre tedavilerini içerir. Hücre tedavisi/kök hücre klinik denemelerinin yakında gerçekleşmesi muhtemeldir.

Floresan anjiyogram; "karanlık koroid" ve sızıntı noktaları görülüyor.
Karşılaştırma için yukarıda fundusun bir fotoğrafı gösterilmiştir.

Fundusun otofloresan çalışmasında karakteristik bir resim, anormal bir lipofuscin birikimi görülür,
aktif ve emilebilir topaklanma tortuları ve RPE atrofisi.
Karşılaştırma için, fundus'un bir fotoğrafı gösterilir (üstte).
 Stargard hastalığı. spektral optik tutarlılık tomografisi(spektral alan optik koherens tomografi - SD-OCT),
Stargard hastalığı. spektral optik tutarlılık tomografisi(spektral alan optik koherens tomografi - SD-OCT), makulanın periferik bölgelerinin retina yapısı nispeten korunurken, makula bölgesinin merkezi bölgesinin retinasının dış katmanlarının arkitektonik kaybı vardır.
Merkezi fossa bölgesinde, retinanın dış katmanlarının tahribatı görülür.
Stargardt hastalığı makulada dejeneratif bir süreci kışkırtır. Kliniği bu patolojiye benzeyen birçok hastalık var. Çeşitli genlerdeki mutasyonlardan kaynaklanırlar. Bu nedenle, hastalık kalıtsal bir patoloji olarak sınıflandırılır.
Ana klinik tezahür hastalık, makulada ve merkezi pigment retinitinde dejeneratif bir süreçtir ve merkezi skotom gelişimi ile görme kaybına neden olur.
Hastalığın özellikleri
Stargardt hastalığı nadir fakat çok ciddi patolojilerden biridir. Genç yaşta kendini gösterir - 6 ila 20 yaş arası 1: 20.000 kişi sıklıkta. Diğer yaş kategorilerinde, kural olarak patoloji oluşmaz. Hastalığın sonuçları felakettir. Tamamen görme kaybı dışlanmaz.
Hastalığın genetik bir temeli vardır. Distrofik süreç maküler bölgeyi etkiler ve pigment epiteli alanından kaynaklanır ve bu da görme kaybına yol açar. Süreç iki yönlüdür.
patoloji formları
Enflamasyon bölgesinin lokalizasyon alanına bağlı olarak patoloji arasında dört tipte açık bir ayrım vardır:
Dejeneratif süreç işaretlenebilir:
- orta periferik bölgede;
- makula bölgesinde;
- parasantral bölgede.
Gözlerin orta kısmında ve çevresinde iltihabın lokalizasyonunu içeren hastalığın karışık bir formu da vardır.
Hastalık gelişim mekanizmaları
Hastalığın nedenleri, 20. yüzyılın ilk yarısında doktor K. Stargardt tarafından tanımlanmıştır. Hastalığa onun adı verilmiştir. Patoloji maküler bölge ile ilgilidir ve bilim adamına göre aynı aile içinde kalıtsaldır. Genellikle, "kırık bronz atrofi" vb. Olarak adlandırılan polimorfik bir oftalmoskopik resim belirtilir.
Konumsal klonlama yoluyla, fotoreseptörlerde en belirgin olana neden olan genin ana lokusu tanımlandı. Bilimde ABCR adını aldı.

Tedavinin temeli, hasta bir kişinin yağ dokusundan alınan kök hücrelerin kullanılmasıdır. terapötik yöntem daha önce bilim adamı V.P. Filatov. Yenilikçi teknoloji sayesinde hastalara kaybettikleri görmelerini geri kazanma ve tam bir yaşam sağlama fırsatı verilir.
Dr. A. D. Romashchenko, biyotıp alanında bir dizi teknolojiyi tescil ettirdi ve aşağıdaki yöntemlerin patentini aldı:
- hastalığın ıslak formunu ortadan kaldırmak için kombine yöntem;
- merkezi ve tapetoretinal distrofinin karmaşık patogenetik tedavisi yöntemi.
Tedavi hangi klinikte yapılır?
En karmaşık hastalığın tedavisi oftalmolojik merkez"O Klinik." Merkez, St. Petersburg gibi bir şehirde bulunuyor. Rusya'da bu teknolojinin kullanıldığı tek merkez olduğu için Stargardt hastalığını sadece bu merkezde tedavi etmek mümkün.

Kök hücre tedavisi güvenli midir?
Uzmanlar, A.D. Romashchenko tarafından geliştirilen teknolojiye göre tedavinin kesinlikle güvenli olduğunu güvenle onaylayabilirler. Terapi için hastanın hücreleri kullanılır, bu da reddedilme olasılığını veya diğer olumsuz sonuçların gelişmesini ortadan kaldırır.
Çözüm
Stargradt hastalığı başladı Erken yaş ve hızla tamamen görme kaybına yol açar. Çok nadir durumlarda, baskın tipe göre kalıtsal olarak alındığında, görme yavaş bir hızda düşer. Hastalara bir göz doktoruna gitmeleri, vitamin almaları ve güneş gözlüğü takmaları tavsiye edilir. Kök hücre tedavisi, patolojiyi ortadan kaldırmanın en etkili yolu olarak kabul edilir.
- makula bölgesinde distrofik değişiklikler ile kendini gösteren ve merkezi görme kaybına yol açan retinanın kalıtsal bir hastalığı. Hastalığın başlangıcı çocukluk veya ergenlik döneminde ortaya çıkar. Hastalar santral skotomlar ve renk görme bozuklukları ile başvururlar. Stargardt hastalığının ilerlemesi tam körlüğe yol açar. Tanı, oftalmoskopi, floresein anjiyografi ve retinal EFI kullanılarak gerçekleştirilir. Tedavi için kullanılır enjeksiyon tedavisi(vitaminler, antioksidanlar, anjiyoprotektörler), fizyoterapi, revaskülarizasyon ameliyatları yapılmakta, otolog doku tedavisi yöntemi geliştirilmektedir.

Genel bilgi
Stargardt hastalığının başka bir adı - juvenil maküler dejenerasyon - hastalığın özünü yansıtır: genç (genç) yaşta başlar ve görsel analizörün reseptör cihazı olan makula hasarı ile karakterize edilir. Hastalık, 20. yüzyılın başında Alman göz doktoru Karl Stargardt tarafından, bir ailede kalıtsal olan gözün maküler bölgesinin konjenital bir lezyonu olarak tanımlandı. Stargardt hastalığının tipik oftalmoskopik belirtileri polimorfiktir: "koroid atrofisi", "boğa gözü", "kırık (dövme) bronz". Patolojinin patojenik adı - “sarı benekli retinal abiyotrofi” - fundus bölgesindeki değişiklikleri yansıtır.
1997'de genetikçiler ABCR geninde bir mutasyon keşfettiler, ihlale neden olmak fotoreseptör hücrelere enerji taşıması gereken bir proteinin üretimi. ATP taşıyıcısının düşüklüğü, retinadaki fotoreseptörlerin ölümüne yol açar. Farklı türde kalıtsal makula dejenerasyonu, göz patolojisi vakalarının %50'sinde görülür. Bunlardan Stargardt hastalığı yaklaşık %7'sini oluşturur. Nosolojik form 1:10.000 sıklıkta teşhis edilir ve ilerleyici bir seyir ile karakterize edilir. Bilateral göz patolojisi genç yaşta (6 ila 21 yaş arası) başlar ve tam görme kaybına kadar ciddi sonuçlara yol açar. Hastalık sosyal bir öneme sahiptir, çünkü genç yaşta sakatlığa yol açar.
Stargardt hastalığının gelişim nedenleri
Miras, hastanın ve ebeveynlerin cinsiyetine bağlı değildir. Patoloji esas olarak otozomal resesif bir tip tarafından iletilir, yani patolojinin kalıtımı cinsiyetle ilişkili değildir (otozomal - cinsiyet dışı kromozomlarla ilişkili) ve her zaman gelecek nesle aktarılmaz (resesif kalıtım). Genetikçilerin son verilerine göre bir genin patolojisi de baskın tipe göre bulaşabilir. ATP taşıyıcı proteinin sentezinin kontrolörü olan gendeki baskın bir kusur kalıtımı türü ile hastalık daha kolay ilerler ve nadiren sakatlığa yol açar. Fundus makulasının makula (üst) reseptör hücrelerinin çoğu çalışıyor. Baskın bir kalıtım tipine sahip hastalarda, hastalık minimum tezahürle ilerler. Hastalar çalışmaya devam edebilir ve hatta araç kullanabilirler.
Makula hücre dejenerasyonunun ana nedeni, enerji eksikliğinden muzdarip olmalarıdır. Gen kusuru, ATP moleküllerini makulanın hücre zarından taşıyan eksik bir proteinin sentezine yol açar - retinanın merkezi, grafik ve renkli görüntünün odaklandığı. Makula alanı yok kan damarları. Koni hücreleri, yakındaki koroidden (koroid) gelen ATP taşıyıcı proteinler tarafından desteklenir. Proteinler, ATP moleküllerini zardan koni hücrelerine taşır.
Normal koşullar altında, fotoreseptör rodopsin, bir ışık fotonu emerek trans-retinal ve opsine dönüşür. Daha sonra trans-retinal, taşıyıcı proteinler tarafından getirilen ATP'nin enerjisinin etkisi altında, opsin ile birleşen retinaya dönüştürülür. Rodopsin bu şekilde geri yüklenir. Bir gen mutasyona uğradığında, kusurlu bir taşıyıcı protein oluşur. Sonuç olarak, rodopsin restorasyonu bozulur ve trans-retinal birikir. Lipofuscin'e dönüşür ve direkt toksik etki koni hücreleri üzerinde.
Stargardt hastalığının sınıflandırılması
Hastalığın türleri, makuladaki hasar bölgesinin prevalansına bağlıdır. Oftalmolojide, aşağıdaki Stargardt hastalığı formları ayırt edilir: merkezi, pericentral, centroperipheral (karma). Merkezi formda, makulanın merkezindeki hücreler etkilenir. Bu, merkezi görme kaybı olarak ifade edilir. Hasta merkezi bir skotom geliştirir (Yunanca "skotos" - karanlıktan). Merkez bölge gözden kayboluyor. Hasta, bakış fiksasyonu noktasında karanlık noktalı bir görüntü görür.
Pericentral form, fiksasyon noktasından uzakta bir skotom görünümü ile karakterize edilir. Bir kişi bakışlarını odaklayabilir, ancak görüş alanının merkezinden yanlardan birinde hilal şeklinde düşmeler fark eder. Zamanla, skotom koyu halka şeklini alır. Merkez-çevresel form merkezden başlar ve hızla çevreye yayılır. Karanlık nokta büyür ve görüş alanını tamamen kaplar.
Stargardt hastalığının belirtileri
Hastalığın belirtileri 6-7 yaşlarında başlar. Kalıtımın türü ne olursa olsun tüm hastalarda merkezi skotom vardır. Uygun bir seyir ile skotomlar görecelidir: hasta net konturlu parlak nesneleri görür ve zayıf renk gamına sahip nesneleri ayırt etmez. Birçok hasta, bir kişinin açık yeşili koyu kırmızı olarak gördüğü kırmızı-yeşil diskromazi tipinin renk görme ihlaline sahiptir. Aynı zamanda, bazı hastalar renk algısındaki değişiklikleri fark etmezler.
Hastalığın ilk aşamasında, periferik görme sınırları değişmez, ilerleme ile merkezi skotomlar genişler ve bu da tam körlüğe yol açar. Merkezi görme kaybının ortaya çıkmasıyla eş zamanlı olarak keskinliği azalır. Stargardt hastalığının son aşamasında optik sinir atrofiler. Kişi görüşünü tamamen kaybeder. Hastalığın hem ilk hem de son aşamalarında diğer organlarda değişiklik yoktur.
Stargardt hastalığının teşhisi
Hastalık başlar çocukluk- Bu, ayırıcı tanı için ana işaretlerden biridir. Oftalmoskopi yardımıyla, karanlık merkezi çevreleyen geniş bir azaltılmış pigmentasyon halkası bulunur. Soluk halkanın çevresinde, bir sonraki hiperpigmente hücre halkası not edilir. Resim "boğa gözü" veya "dövme bronz"u andırıyor. Foveolar refleks negatif. Makula elevasyonu tanımlanmamıştır. Makulayı incelerken, çeşitli boyut ve konfigürasyonlarda sarımsı beyaz lekeler not edilir. Zamanla, kapanımların sınırları bulanıklaşır, lekeler gri bir renk alır veya tamamen kaybolur.
Stangardt hastalığında perimetri sırasında, pozitif veya negatif (hasta onları hissetmez) merkezi skotomlar not edilir. Hastalığın merkezi formu ile kırmızı-yeşil döteranopi gelişir. Çevresel form, renk algısının ihlali ile karakterize edilmez. Mekansal kontrast duyarlılığı tüm aralıkta değişir: yüksek frekans bölgesinde (6-10 dereceye kadar orta bölgede) yoktur ve orta frekans bölgesinde azalır.
Hastalığın ilk aşamasında, merkezi distrofi formunda maküler elektrografide bir azalma vardır. Daha fazla ilerleme ile elektrik potansiyelleri kaydedilmez. Distrofi orta periferik bölgede bulunduğunda, ilk aşamada normal elektrografi ve elektrookülografi not edilir. Daha sonra elektroretinografinin koni ve çubuk bileşenlerinin değerleri normalin altına düşürülür. Hastalık asemptomatiktir - görme keskinliği ve renk algısı bozulmadan. Görme alanının sınırları normal aralıktadır. Karanlık adaptasyon biraz azalır.
"Boğa gözü" arka planına karşı floresan anjiyografi yardımı ile hipofloresan bölgeleri tespit edilmez, kılcal damarlar, "sessiz" veya "karanlık" koroid görülür. Atrofi alanlarında, retina pigment epitel hücrelerinin hiperfloresan alanları görülebilir. Fundusun merkezi bölgesindeki histolojik inceleme şunları belirler: artan miktar pigment - lipofuscin. Hipertrofik ve atrofik pigment epitel hücrelerinin bir kombinasyonu vardır.
Moleküler genetik analiz, hastalığın belirtilerinin başlangıcından önce bir gen mutasyonunu fark etmenizi sağlar. Nükleotid ikamelerini saptamak için, birkaç DNA probu - "moleküler işaretçiler" kullanılarak gerçek zamanlı PCR gerçekleştirilir. Stargardt hastalığının ayırıcı tanısı, edinilmiş ilaca bağlı distrofiler, Kandori retina lekeleri, ailesel drusen, juvenil retinoskizis, baskın ilerleyici foveal, koni, koni-çubuk ve çubuk-koni distrofisi ile gerçekleştirilir.
Stargardt hastalığının tedavisi ve prognozu
Etiyolojik tedavi yoktur. Genel bir yardımcı tedavi olarak, taurin ve antioksidanların parabulbar enjeksiyonları kullanılır, giriş vazodilatörler(pentoksifilin, bir nikotinik asit), steroid ilaçlar. Vitamin tedavisi kan damarlarını güçlendirmek ve kan akışını iyileştirmek için yapılır (vit. B, A, C, E grupları). Fizyoterapötik tedavi yöntemleri gösterilmiştir: ilaç elektroforezi, ultrason, retina lazer stimülasyonu. Retinanın revaskülarizasyon yöntemi, makula bölgesine bir kas lifi demeti nakledilerek kullanılır. Hastanın yağ dokusundan alınan kök hücreler kullanılarak otolog doku tedavisinin patogenetik rejeneratif oftalmik teknolojisi geliştirilmektedir.
Stargardt hastalığı erken yaşta başlar ve hızla görme bozukluğuna yol açar. Nadir durumlarda, baskın bir kalıtım türü ile görme yavaş yavaş düşer. Hastaların bir göz doktoruna görünmeleri önerilir vitamin kompleksleri ve güneş gözlüğü takıyor.
Juvenil makula dejenerasyonu veya Stargardt hastalığı, retinanın kalıtsal bir makula dejenerasyonu türüdür. Hastalık 12-20 yaşlarında tespit edilir ve her iki gözde görme keskinliğinde ilerleyici bir azalma ile kendini gösterir.
Patolojinin konumuna bağlı olarak, 4 tür jüvenil maküler dejenerasyon ayırt etmek gelenekseldir:
- Makula bölgesinde;
- Orta çevrede;
- Parasantral bölgede;
- Merkezi ve çevresel bölgelerde (karışık form).
Halen devam etmekte olan genetik çalışmalar, juvenil makula dejenerasyonu ve Franceschetti hastalığının (sarı benekli fundus) aynı hastalığın fenotipik özellikleri olduğunu kanıtlamaktadır.
nedenler
Hastalık otozomal resesif bir kalıtım yoluyla bulaşır, nadiren otozomal baskındır. Jüvenil maküler dejenerasyon için pozisyonel klonlama ile tanımlanan majör lokus hastalığa neden olan gen. Fotoreseptörlerde ifade edilir ve ABCR olarak adlandırılır. ABCR, sözde süper ailenin bir üyesidir. ATP bağlayıcı kaset taşıyıcı, dizi olarak insan RmP geni ile aynıdır.
Hastalığın otozomal dominant kalıtımı ile, mutasyon genlerinin 13q kromozomlarında ve 6q14'te lokalizasyonu belirlendi. ABCR mutasyonlarının, yaş ve koni-çubuk dejenerasyonu ile ilişkili eksüdatif olmayan maküler dejenerasyon formu olan bir hasta alt popülasyonunda mevcut olduğu ortaya çıktı, bu da hastaların kan akrabalarında AMD geliştirmenin genetik olarak belirlenmiş bir riskini ortaya koyuyor.
Uzmanımızın hastalıkla ilgili videosu
Hastalığın belirtileri
Pigment epitelinde retina gözlerde yoğun bir lipofuscin birikimi var. Sürece, pigment epitel hücrelerinde pH'da bir artışla birlikte lizozomların oksidatif fonksiyonunun zayıflaması eşlik eder ve bu da membran bütünlüğünde bir değişikliğe yol açar.
Jüvenil distrofinin merkezi formu ile hastalık geliştikçe, makula bölgesinin oftalmoskopik resmi sonraki görünüm: "kırık metal", sonra "boğa gözü", ardından "dövme bronz" ve sonuç olarak koroid atrofisi.
"Boğa gözü" fenomeni aşamasında oftalmoskopi, geniş bir hipopigmentasyon halkası ve onu takip eden başka bir hiperpigmentasyon halkası ile çevrili karanlık bir merkez ortaya koymaktadır. Retina damarları değişmez, optik disk, papillomaküler demetin sinir liflerinin atrofisine bağlı olarak temporal tarafta soluktur. Makula elevasyonu gibi foveolar refleks de yoktur.
Gözün arka kutbunun retina pigment epitelinde sarımsı beyaz lekelerin varlığı bulunur. Noktaların farklı bir boyutu, şekli ve konfigürasyonu vardır - bu en çok karakteristik semptom sarı benekli fundus. Zamanla lekelerin şekli, rengi ve boyutu değişebilir. Başlangıçta açıkça tanımlanmış kenarları olan sarımsı lekeler, genellikle birkaç yıl sonra gri hale gelir, sınırları bulaşır veya kaybolur.

Hastalığın teşhisi
Bir anamnez toplama sürecinde, tanıda önemli bir rol oynayan hastalığın başlangıç zamanı (tezahür yaşı) ortaya çıkar.
Laboratuvar ile histolojik çalışmalar fundusun orta bölgesinde, pigmentte bir artış, bitişik retina pigment epitelinin atrofisi, kombine atrofi ve pigment epitelinin hipertrofisi not edilir. Lipofuscin benzeri malzeme ile sarı lekelerin temsili.
Sırasında araçsal araştırma Jüvenil makula dejenerasyonu olan hastalarda perimetri ile, sürecin zamanlamasına ve prevalansına bağlı olarak erken çocukluk veya ergenlikten itibaren farklı boyutlarda nispi veya mutlak merkezi skotomlar bulunur. Sarı benekli fundus durumunda makulada herhangi bir değişiklik olmaz, görüş alanı genellikle değişmez.
Patolojik sürecin merkezi lokalizasyonu olan çoğu hastada renk anomalileri, genellikle daha belirgin olan deuteranopia veya kırmızı-yeşil diskromazi olarak gelişir.
Sarı benekli bir fundus durumunda renk görüşü değişmeyebilir. Mekansal kontrast duyarlılığı, tüm uzamsal frekans aralıklarında önemli ölçüde değişir, orta bölgede önemli ölçüde azalır ve yüksek uzamsal frekanslar bölgesinde (desen-koni distrofisi olarak adlandırılır) tamamen yoktur. Retinanın merkez bölgesinde 6-10 derece arasında kontrast duyarlılığı yoktur.
Görme keskinliği, görme alanı ve renkli görme normaldir. Karanlık adaptasyon genellikle normaldir veya biraz azalır.
FAG'da, normal bir arka plana sahip tipik bir "boğa gözü" fenomeni durumunda, "yokluk" bölgeleri veya bazı durumlarda gynofloresan, görünür koryokapillerlerin yanı sıra "karanlık" veya "sessiz" varlığı ile ortaya çıkar. " koroid. Makula bölgesinde floresan olmaması, floreseini koruyan lipofuskin birikimi ile açıklanır. Hipofloresanlı alanlar bazen pigment epitel tabakasının atrofi alanlarına göre hiperfloresan hale gelir.
Ayırıcı tanı
Makuladaki birçok dejeneratif hastalığın klinik tablosunun benzerliği tanıyı ciddi şekilde engellemektedir. Juvenil makula dejenerasyonunun ayırıcı tanısı ailesel drusen, Kandori retina lekeleri, progresif dominant foveal distrofi ile yapılır; jüvenil retinoskizis; koni, koni-çubuk, çubuk-koni distrofileri; vitelliform makula dejenerasyonu; tıbbi distrofiler.
Tedavi ve prognoz
Bugüne kadar, jüvenil makula dejenerasyonu için patojenetik olarak doğrulanmış bir tedavi yoktur. Bir uzmanı sürekli izlemek, görüş alanını kontrol etmek, ERG, EOG'yi izlemek gerekir.